2 Pre-processing
2.1 Quality Control of Raw Reads
We obtain an overview of the quality of the raw reads with FastQC.
module load fastqc/0.11.9
fastqc -t 6 AJ_GIAB_fastq/HG002_1.fastq AJ_GIAB_fastq/HG002_2.fastq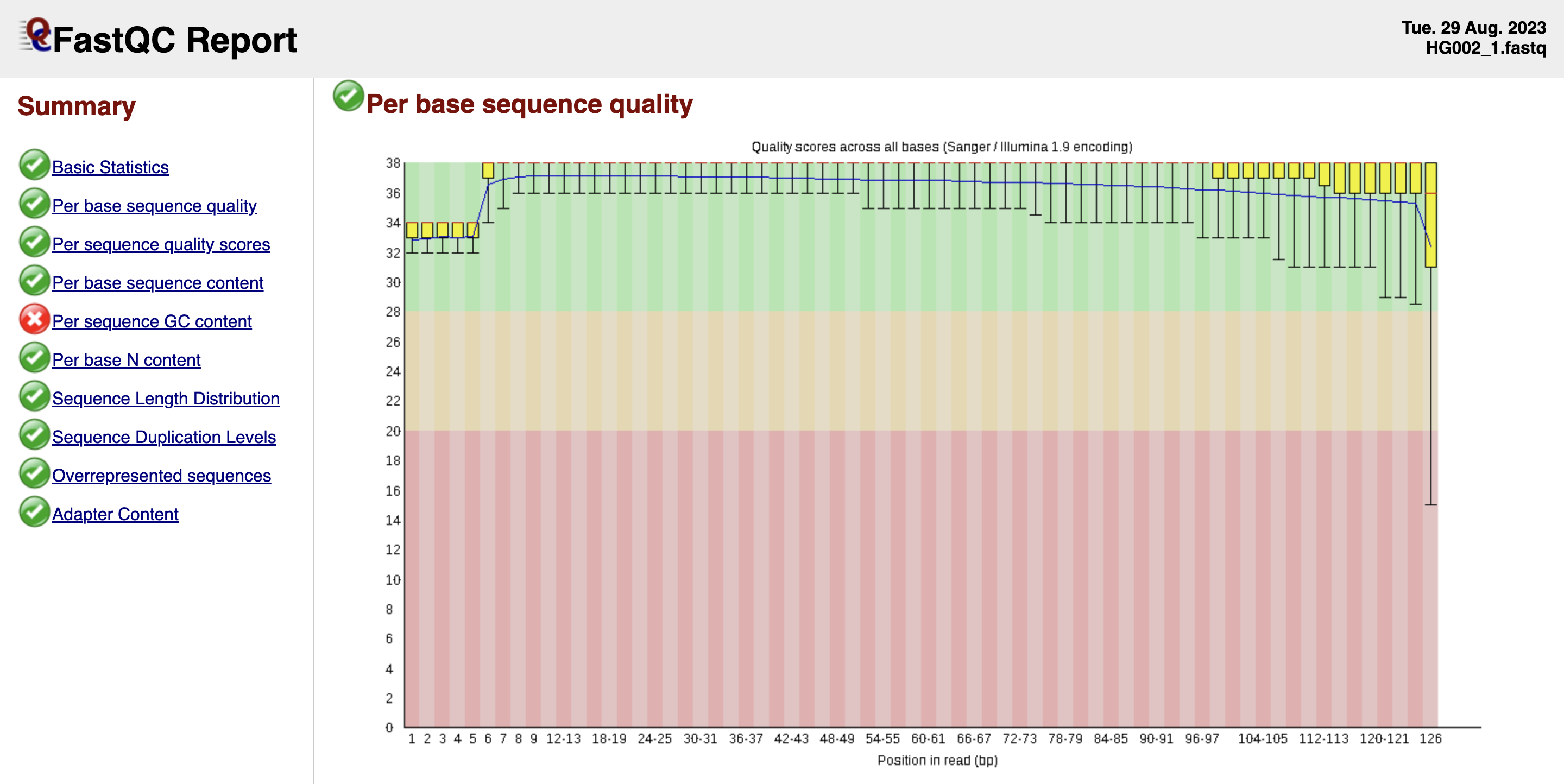
FastQC report for HG002 paired end R1.
2.2 Alignment
We trim raw reads with fastp, then map reads to the GRCh38 reference genome with the BWA-MEM algorithm and convert SAM to BAM with samtools.
module load fastp/0.23.4
module load bwa
module load samtools
export reference=$MUGQIC_INSTALL_HOME/genomes/species/Homo_sapiens.GRCh38/
id=""
sm="HG002"
genome/bwa_index/Homo_sapiens.GRCh38.fa
fastp -i HG002_1.fq.gz -I HG002_2.fq.gz \
--stdout --thread 2 \
-j "fastp-HG002.json" \
-h "fastp-HG002.html" \
2> "fastp-HG002.log" \
| bwa mem -v 2 -M -t 32 -p \
-R "@RG\tID:$id\tPL:ILLUMINA\tLB:$id"_"$sm\tSM:$sm" \
$reference - 2> "bwa-HG002.log" \
| samtools view -@ 16 \
-O BAM \
-o "aligned_HG002.bam" \
2> "samtools-HG002.log"2.3 Mark Duplicates
We identify read pairs that are likely to have originated from duplicates of the same DNA fragment with GATK MarkDuplicates.
export GATK_JAR=/cvmfs/soft.mugqic/CentOS6/software/GenomeAnalysisTK/GenomeAnalysisTK-4.1.8.1/gatk-package-4.1.8.1-local.jar
java -Xms60G -Xmx60G -jar $GATK_JAR MarkDuplicatesSpark \
-I aligned_HG002.bam \
-O aligned_HG002_markdup.bam \
--spark-master local[12]2.4 Base Quality Score Recalibration
We use BQSR to recalibrate the base quality of reads based on on various covariates, i.e., read group, reported quality score, machine cycle, and nucleotide context.
java -Xms4G -Xmx4G -jar $GATK_JAR BaseRecalibrator \
-I aligned_HG002_markdup.bam \
-R $reference \
-O aligned_HG002_markdup_bqsr.report \
--known-sites BQSR/Homo_sapiens_assembly38.dbsnp138.vcf \
--known-sites BQSR/Homo_sapiens_assembly38.known_indels.vcf.gz \
--known-sites BQSR/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
java -Xms2G -Xmx2G -jar $GATK_JAR ApplyBQSR \
-I aligned_HG002_markdup.bam \
-R $reference \
--bqsr-recal-file aligned_HG002_markdup_bqsr.report \
-O aligned_HG002_markdup_bqsr.bam2.5 Quality Control of Alignment
samtools stats collects statistics from BAM files and outputs in a text format.
samtools stats aligned_HG002_markdup_bqsr.bam > aligned_HG002_markdup_bqsr_stats.txtMosdepth summarizes basic statistics of the alignment (number of reads, coverage, GC-content, etc.) and produces a number of useful graphs.
module load mugqic/mosdepth/0.3.4
mosdepth --by $exome HG002_mosdepth_output aligned_HG002_markdup_bqsr.bamMultiQC generates a single HTML report summarizing all samples in the project.
module load multiqc
multiqc .